


Treating cancer: Progress on many fronts
In rich countries half of cancers are now survivable. And better understanding means that more cures are coming, says Natasha Loder.
THERE are few whose lives have not been touched by cancer. It cuts down friends, loved ones, siblings, spouses, parents and children. And it does so more than it used to. A generation ago, one in three people in the rich world could expect one day to hear the fateful words, “I’m afraid you have cancer.” In some countries it is now approaching one in two. The longer other things do not kill you, the more of the wear and tear that leads to cancer your cells accumulate. Live long enough and it will be the reward.
Worldwide, cancer is the second leading cause of death after heart disease; it killed 8.8m people in 2015, three-quarters of them in low- and middle-income countries. Between 2005 and 2015 the number of cases increased by 33%, mostly owing to the combined effects of ageing and population growth. New cases are expected to increase by 70% in the next 20 years.

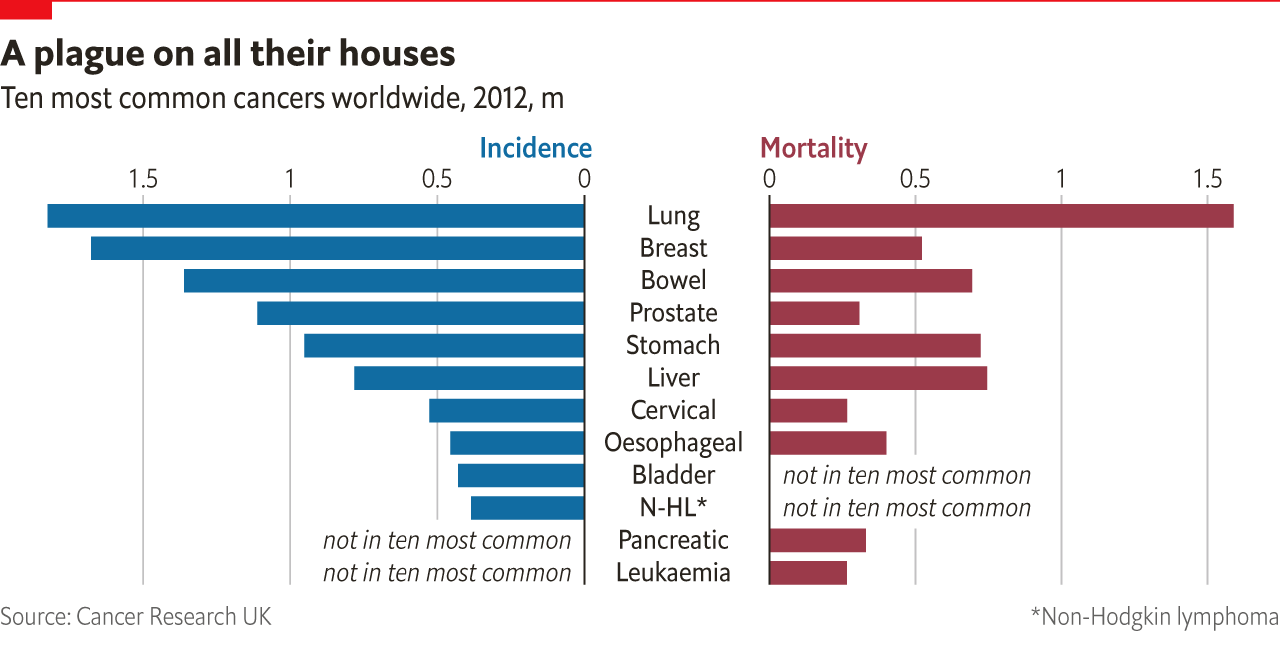
Set against this rise is the fact that, in rich countries, cancer is becoming more survivable. Today 67% of patients in America will survive for at least five years. Different cancers fare differently, as do different sorts of patients—cancer has proved more treatable in children than in adults. Some cancers, such as that of the pancreas, have seen barely any improvement. But there are general grounds for optimism.
New research tools, such as easily generated antibodies, rapid gene sequencing and ever easier genetic engineering, have revolutionised biologists’ understanding of cancer. This understanding has allowed more specific approaches to the disease to be developed, and the trend will continue. What is more, the tools of molecular biology have moved out of the lab and into the clinic. Genetic tests are used to find the precise vulnerabilities of a particular patient’s cancer. Antibodies attack the specific molecules that have gone haywire. The cells of patients with cancer are engineered to better fight the disease.

And in the current decade a whole new branch of therapy has sprung up. Unshackling the immune system’s response to cancer, once a pipe dream, has become practical medicine, with approved therapies for eight kinds of cancer. The excitement at oncology conferences is palpable.
As these advances have arrived regulators have increased the speed with which treatments for life-threatening diseases are approved. This is in some ways a mixed blessing—some expensive drugs with little if any benefit are nevertheless getting to market. But it has encouraged an unprecedented wave of investment and innovation. The pipeline of oncology drugs in clinical development has grown 45% in the past decade. There are currently about 600 drugs under development at biotech and pharma companies.
The picture is not uniformly rosy. In both treatment and prevention the poor are ill served. Basic chemotherapy and pain relief is difficult to come by in many parts of the world. The failures are not limited to poor countries. Cancers due to bad diet, obesity, alcohol abuse and smoking could all be reduced a great deal in wealthy ones. And while vaccination against human papilloma virus is routine in Rwanda, it is still limited in America—which means thousands of American women will face cervical cancers they could have avoided in years to come.
But if some low-hanging fruit still go tragically, lethally unpicked, progress is not merely possible. It is happening.
It all starts with a single cell
DIANE MILLEY, a teacher, remembers getting the small, dry cough just before school broke up for the summer in 2013. She wasn’t worried: she considered herself generally healthy—she ran three times a week and went to the gym. Her doctor in Bradford, Massachusetts, put her on a course of antibiotics. When they didn’t work she had an X-ray. It showed nodules across her lungs. A bronchoscopy was ordered to retrieve a tissue sample from her lung. As she came round from the anaesthetic she remembers overhearing two medical staff talking. One said “It’s malignant.” She had late-stage lung cancer.
Ms Milley’s body, like all human bodies, contained tens of trillions of copies of her genome. In theory, all those copies should be more or less the same. In practice, over the years, they all get knocked around in different ways. The oxygen that powers cell metabolism damages the DNA on which the genes are stored as a matter of course; so do background radiation and exposure to the many low-level carcinogens; so do sunlight and infection with viruses; so do choices about diet and recreational drugs, notably alcohol and tobacco (from which Ms Milley abstained).
The vast bulk of this damage is quickly fixed by DNA-repair enzymes; fewer than one mutation in a thousand persists. But wear and tear builds up. Many such changes make little or no difference. A few will be of consequence to the cell concerned, reducing or eliminating its capacity to do its job. But the loss of a single cell’s contribution matters not a jot.
There are some genes, though, where uncorrected damage can matter a lot. Foremost are the genes which control cell growth, such as HER2, which tells the cell how to make a protein called human epidermal-growth-factor receptor type 2. This is a protein that, when it sees a particular hormone, tells the cell it is in to divide. Mutations in the HER2 gene can make cells proliferate when there is no need. When they do so their daughter cells, which will share that HER2 mutation, will go on to do the same.
Among some 20,000 genes in the genome there are dozens which, like HER2, can cause unwanted cell division when they go wrong. To forestall such problems there are various tumour-suppressor genes whose job is to make sure that cells damaged in this way shut themselves down. The best known is the gene for a protein called p53, which stops cells from reproducing if their DNA is damaged. But these tumour-suppressor genes, too, are subject to mutation.
The numbers game
Thus over time, as genetic damage accumulates, the likelihood rises that somewhere in the body’s trillions of cells there is one that has, through five or six mutations in key genes, developed the ability to grow without check. This likelihood is not the same for everyone. Some people start off with quirks in their genome which make them more susceptible. Take the genes BRCA1 and BRCA2, which describe proteins that repair DNA; people who inherit a damaged version of one or the other face a higher risk of cancer (in particular, breast and ovarian cancer) because, with one crucial function already compromised, it takes fewer mutations for a tumour to get going.
Once a cancer has begun its unruly growth it will pick up more and more mutations: the cancer genome project at the Sanger Institute, outside Cambridge in England, has found that cancers can have as few as ten mutations or as many as a few hundred. Though all the cells in the cancer are descended from one parent cell, they become increasingly diverse over time. Some cells come loose and start new tumours of their own elsewhere. The body’s immune system will often recognise that something is amiss and try to fight the cancer and slow its spread. Sometimes it wins, stopping the cancer or killing it. Sometimes it doesn’t.
When Ms Milley’s cancer was diagnosed all the things that could go wrong already had; the tumour was well developed and had spread through the lung and beyond. It would have been far better for her if it had been diagnosed earlier (see chart). But with lung cancer, as with many other forms of the disease, there are often few symptoms until the disease is already at an advanced stage. If cancer could be reliably detected earlier, many lives might be saved.


In some wealthy countries, some cancers—for example, those of the breast, prostate and cervix—are regularly sought out before they start to cause symptoms. Now researchers are trying to improve diagnostic tools even further, so that more types of cancer can be found early on (and with greater reliability). For some it is a terribly personal hunt. Billy Boyle, the president of a small biotech company, Owlstone Medical, based in Cambridge, in England, is one of them. He lost his wife Kate, mother to their two young boys, on Christmas morning in 2014. She died of colon cancer that had been picked up too late. Mr Boyle says that if colorectal cancer is detected early, 95% of sufferers survive. Only 6% survive if the cancer reaches stage four. For many cancers, early detection is “our greatest opportunity to improve survival,” says Mr Boyle.
Mr Boyle wants to detect cancer on the breath using an ion-mobility spectrometer—a gadget that weighs chemicals by passing them through an oscillating electric field. The breath contains a wide range of organic molecules that reflect what is going on in the body’s metabolism. Cancers, which affect the metabolism, should in so doing change the pattern of molecules on the breath. Although Owlstone’s system is very small—it fits on a chip the size of a coin—it is sensitive, identifying molecules at a level of a few parts per billion. The firm hopes that when it has identified molecular “fingerprints” associated with particular cancers it will be able to detect the disease earlier than other tests do.
Improved diagnostics can do more than pick up cancers sooner. They can also reveal the cancers’ weaknesses. Because cancer drugs work in different ways, some will do well against a tumour with one set of mutations but leave unscathed one that has become cancerous by some other pathway. Troy Cox, head of Foundation Medicine, a diagnostics company based in Cambridge, Massachusetts, says that in America 14 cancer drugs now have “companion diagnostics”—tests that show whether a cancer is likely to be susceptible to them or not. Ms Milley’s lung cancer, for example, turned out to harbour a mutation which meant she could be treated with a drug that targets that specific protein (see next section).
So far, such genetic tests are used when planning therapy for 50% to 60% of solid tumours, according to Foundation. New drugs, new understanding of cancer mechanisms and new technologies that can scan many genes for mutations at once mean such testing will be more informative in the near future. Many, including England’s chief medical officer, Sally Davies, want cancer patients to be routinely offered genetic screening of their tumours. Foundation and ThermoFisher, a diagnostics firm in Waltham, Massachusetts, are hoping to encourage this by offering every gene of interest on a mass-produced chip. Some of these mutations will help doctors pick the best drugs for that particular cancer, others may indicate how it is likely to develop. The tests would also identify mutations for which there is not yet an approved therapy—but for which there is one in clinical trials.
David Hyman, at the Memorial Sloan Kettering Cancer Centre in New York, worked on a trial of an experimental drug, larotrectinib, that was expected to work in cancers where a gene called NTRK1 had undergone a specific mutation. Because that mutation is found in less than 1% of all cancer patients, recruiting people to the trial was a “Herculean effort”, he says. It was worth it, though. The drug was tested on 50 patients with 17 different types of tumour. In results published in June, 78% of patients with 12 different tumour types responded to the drug.
Aside from picking the right drugs, genetic tests are also starting to reveal more about the outcome and risks of any individual cancer—something that is useful for deciding whether a cancer needs to be treated at all. The MammaPrint test, made by Agendia, based in Amsterdam, analyses the activity of genes in early-stage breast cancer. If women with early-stage breast cancer were routinely tested in this way, those who will not need chemotherapy after surgery could be picked out (a recent study of patients found 58% to be in this category). A similar test is available for prostate cancer from the firm Myriad Genetics, based in Salt Lake City, Utah. A recent study suggests that people who have inherited a mutation in the P53 tumour-suppressor gene might be well advised to have whole-body MRI scans to screen for cancers, since their unsafeguarded cells are at particular risk.
Identifying genes from tumours normally means retrieving cancer cells via biopsies. This is invasive and often done only once in the course of the disease. But cancers are both heterogeneous and labile; elsewhere in a tumour, and later in a tumour’s progression, things may look different.
These challenges are now being tackled with blood tests, a technique termed “liquid biopsy”. Tumours shed DNA into the blood, and these circulating fragments of DNA can be tested for mutations. Regularly testing this DNA could be a way of keeping track of a tumour’s mutations. The Institute for Cancer Research, based in London, recently showed that it could use a liquid biopsy to pick out whether a patient was likely to benefit from a new type of drug called a PARP inhibitor. Using liquid biopsies the researchers were able to find out if the drug was doing any good in just four to eight weeks. Liquid biopsies are also a promising technology for the routine monitoring of patients who have been successfully treated for cancer, lest their disease return. Mark Roschewski, a researcher with America’s National Cancer Institute, the NCI, thinks the technology could be “orders of magnitude more sensitive than radiographic imaging”.
Biopsies optimised
The big question for the firms developing these liquid biopsies is whether the technology will also be suitable for the early-detection market that Mr Boyle is chasing with his breath tests. Guardant Health, a firm based in Redwood City, California, currently offers a liquid biopsy that allows patients to obtain a genetic profile of their tumour. It is using the data it gathers to look at the feasibility of early detection. Helmy Eltoukhy of Guardant says the firm is “agnostic” about the markers it seeks in the blood, meaning that its researchers will not look just for DNA from tumours—if the data suggest that RNA (a relative of DNA) or proteins provide the telltale fingerprint, then that is what they will look at.
All diagnostic tests have to overcome two hurdles. They have to be sensitive enough to identify those who have the disease correctly and also specific enough that they do not see signs of the disease when it isn’t actually present. The more widely they are used, the more important that second requirement gets; false positives are a pervasive problem with existing tests such as mammograms and PSA, a test for prostate cancer. (This is why PSA screening, while common in America, is much less prevalent in Europe.)
In liquid biopsies the challenge will be to detect cancer-specific signals against a noisy and confusing background. Barry Kramer, director of the division of cancer prevention at the NCI, warns that the same marker can have different functions in different organs. He notes that a programme screening infants for neuroblastoma was halted after it started to pick up too many growths that did not merit clinical concern and didn’t reduce the death rate. Specificity, says Mr Eltoukhy, is early detection’s Achilles’ heel. Others warn that liquid biopsies aimed at DNA will never be sensitive enough for early detection, because early tumours may shed very little DNA, or shed it only occasionally; other molecules might prove more telling.
Nonetheless, biotech is gung-ho about the idea. Grail, a liquid-biopsy startup in Silicon Valley spun out of Illumina, a sequencing firm, recently raised $900m. Earlier this year Guardant raised $360m, and Alphabet invested $65m in Freenome, a San Francisco startup with similar plans. Grail has begun a trial of its technology which will enroll 120,000 women who are receiving mammograms to see if its technology really does offer early detection.
Whether it will make sense to adapt liquid biopsies to population screening will depend on their costs—currently still too high for widespread use—their sensitivity and, crucially, their false-positive rates. Unnecessary investigations after false positives are both worrying and debilitating for patients and costly for the health-care system. But some, such as Luis Diaz, an oncologist at Memorial Sloan Kettering, argue that initial overdiagnosis is a necessary part of moving ahead: “One never learns to ride a bike without falling off.”
The costs and difficulties of blood screening are one of the things that tiny Owlstone has going for it. Testing the breath for metabolites doesn’t require the tumours to have started shedding DNA. Britain’s NHS is running a £1.1m trial of the technology in patients suspected of having lung cancer who are also being examined by other means. If this finds the technology to be reliable it might be expanded for use in population screening. In July Owlstone said it would collaborate with academic partners to see if breath biopsies could be expanded to pick up bladder, breast, kidney, pancreatic, prostate, brain, and head and neck cancers.
There is no question that blood biopsies will be at the heart of the future of tracking and profiling tumours. But for early detection other options might yet win out; success will not hinge on which company starts with the most money but which offers the biggest bang for the buck. Health-care systems will seek to adopt technologies that work at scale. The benefit will be that more cancers can be cured with the most basic, oldest and most effective methods of cancer treatment.

Surgery, radiotherapy and cancer drugs are all becoming more tightly focused
MS MILLEY’S primary tumour was in the middle lobe of her right lung, which surgeons removed entirely. Surgery is an ancient form of cancer treatment and still a common one. Today’s surgeons have everything from lasers to cryosurgery—the freezing of abnormal tissue—at their disposal. By and large, they use this expanding range of tools to cut out less and less. Ultrasound, magnetic-resonance imaging (MRI), X-ray tomography and positron-emission tomography (PET) scans have between them eliminated much of the need for “exploratory surgery” to understand the scope of a cancer.
Often surgery goes hand-in-hand with radiation therapy. Soon after the discovery of X-rays at the end of the 19th century it became clear that radiation which killed cells could be used as a cancer therapy. In its early days practitioners judged the correct dose by trying their machines out on their own arms, looking for a pink reaction on their skin. Many went on to develop leukaemia.
Today radiotherapy is considerably safer for its practitioners and more beneficial to its recipients. After a cancer is cut out, radiation is frequently used to kill the cancer cells the surgeon’s knife has missed. It is also sometimes used to destroy the tumours themselves, particularly in places where surgery would be hard. In rich countries about half of patients with localised cancers receive radiotherapy. Two out of five of those treated for cancer and cured in Britain will have had treatment which consisted of radiotherapy either alone or in part. Breast and prostate cancers respond well to it.
To make all this possible, medical physicists produce beams of X-rays, gamma radiation, neutrons and, increasingly, protons; they have ever more sophisticated ways of ensuring that these cell-damaging energies are delivered to the tumours being targeted, rather than to healthy tissue nearby. Ms Milley experienced this when she had a superficial brain metastasis dealt with by stereotactic radio-surgery. The procedure uses 3D imaging to determine the exact location of a tumour, at which point a number of different beams are focused on it from various directions (see diagram on next page). The idea is that only in the part of the brain where all the beams cross is the dose high enough to kill cells—the individual beams, on their way in and out, do comparatively little damage. The idea is to match the extent of the lethal criss-crossing as closely as possible to the location of the tumour. It is a way of achieving what Emma Harris, a medical physicist with the Institute of Cancer Research (ICR) in London, calls the current state of the art: “Shaping the beam and varying the intensity of the radiation dose to create exquisite volumes of radiation.”

Proton therapy offers another way to deal death to tumours while sparing the surrounding tissues. By choosing the right energy for the beam physicists can determine how deep into the tissue it will get before doing most of its damage. This specificity is seen as particularly useful in tumours that are near eyes, brains and spinal cords.
Radiation can also be emitted inside the body; radioactive pellets and seeds can be put right where they are needed. A new version of this approach is being developed by Nanobiotix, a biotech firm based in Paris, which is developing nanoparticles containing hafnium oxide which generate electrons when exposed to X-rays. When these nanoparticles are injected into tumours that are then zapped with X-rays they increase the damage done.
As well as surgery on the lung and radiation treatment for the tumour in her brain, Ms Milley also had chemotherapy—the third of the 20th century’s medical responses to cancer. She was given a cocktail of cisplatin, a drug containing platinum that was approved in 1978, and Alimta (pemetrexed).
Chemotherapy’s origins can be traced back to the development of chemical weapons in the first world war. Looking into the records of soldiers affected by mustard gas, two doctors at Yale University, Louis Goodman and Alfred Gilman, noticed that many were short of white blood cells. They wondered if this meant that cancers in which white blood cells proliferate—lymphomas—might be treated with something similar. The first patient to receive this treatment was a man with advanced lymphoma who is known today by the initials “J.D.”. His symptoms were greatly relieved.
The treatment worked because mustard gas damages cells’ DNA, stopping cell division. These effects are not specific to cancer cells; but because cancer cells divide a lot, such poisons are particularly bad for them. In 1947 aminopterin, a chemical which messes up cell division by interrupting the metabolism of folic acid, was found to produce remissions in children with acute leukaemia. This drug was a precursor to methotrexate, a treatment which provided the first cures of a metastatic cancer in 1956 and is still commonly used today. By the 1960s, chemotherapy had induced long-term remissions, and even cures, of Hodgkin disease, a lymphoma, and childhood acute lymphoblastic leukaemia. Cures of testicular cancer arrived in the 1970s. Though few cancers can be cured with chemotherapy on its own, many can be set back a long way and controlled for quite some time. Chemotherapies, like radiation therapies, are often used to mop up the cancer left over when primary tumours have been excised.
Elective affinities
One problem with chemotherapy is that cancers can become resistant to it. Most cancers are genetically heterogeneous, because the cells accumulate new mutations as they grow. Some of these mutations can make the cells less susceptible to the chemotherapy. As treatment continues, such cells become more numerous, and as they divide they go on to accumulate mutations that make them even more resistant—the cancer evolves resistance to chemotherapy rather as an infection can evolve resistance to antibiotics. This is why chemotherapies are now often used in combination; it is harder to evolve resistance to two or three drugs at the same time.
Another problem with chemotherapy is that it attacks cells that are dividing for perfectly legitimate non-cancerous reasons, too. Hence the side-effects, which include fatigue, hair loss, mood changes and nausea. The severity of the effects vary greatly from person to person, and some, such as nausea, can be treated with secondary drugs under some circumstances. But some chemotherapies can have long-term side-effects, damaging the heart, the nerves and fertility.
Before taking on her chemotherapy, though, Ms Milley was given another treatment: Tarceva (erlotinib). Tarceva is a small molecule which disrupts signals transmitted by a protein called the epidermal growth-factor receptor (EGFR). At least eight mutations that cause the EGFR to be constantly active have been tied to lung cancer, and Ms Milley had one of them. Her course of Tarceva saw all the tiny tumours across her body shrink, one by 60%. She went back to work.
A key tool for targeting cancer-specific pathways and molecules is the antibody. Antibodies are proteins made by the immune system which stick to a particular bit—the “antigen”—of a particular molecule. Turning them into mass-produced drugs has been one of the biotech industry’s triumphs. In the 1990s they started to come into use as cancer therapies. Aimed at antigens that crop up on cancers, but not other cells, they are far more specific than older chemotherapies. Rituxan (rituximab), an antibody which targets a protein on the surface of the immune system’s B-cells that misbehaves in B-cell non-Hodgkin lymphoma, was approved 20 years ago, in 1997. Other early targeted therapies that blocked growth signals in different cancers included Herceptin (trastuzumab) and Erbitux (cetuximab), which are both antibodies, and Iressa (gefitinib) and Gleevec (imatinib), which are smaller molecules like Tarceva. These drugs transformed the treatment of many cancers. Herceptin, for example, dramatically altered the outcome of breast cancer in patients with the HER2 mutation. With Herceptin as part of a two-drug therapy, a woman diagnosed with the metastatic form of the disease can hope to survive for almost five years; previously it was 20 months.
Another promising targeted approach involved aiming drugs at the creation of new blood vessels. If tumours are to grow beyond a few millimetres in size they need to encourage new blood vessels to bring them nutrients. Drugs which inhibit this process arrived in 2004 with Avastin (bevacizumab). It is currently used to treat advanced colorectal, kidney and lung cancers.
A third approach attacks DNA repair systems. Losing some of the ability to repair DNA helps cancers accumulate mutations, and is often part of how they get started. But the cancers need to keep some residual DNA repair functions; otherwise the cells will simply die. Thus cancers that have mutations in the BRCA1 and BRCA2 genes rely heavily on a backup DNA repair mechanism which uses proteins called poly-ADP-ribose polymerases (PARPs). Now targeted drugs have been designed to inhibit this repair mechanism. In its absence, massive genetic damage drives cancer cells to their death. Some of these PARP inhibitors have been shown to help in BRCA-linked breast cancers, and there are promising results in ovarian cancer. They seem also to have promise in some prostate and pancreatic cancers.
Finding targets for such therapies has been made far easier by the sequencing of the human genome and the remarkable reductions in the cost of sequencing DNA which followed on from it. With a baseline genome for comparison, identifying the mutations in cancers became much easier. Once found, these genes can be used to understand the molecular workings of the disease and, in theory, to find new targets for drug developers.
Imperfect chemistries
Mike Stratton, director of the Sanger Institute, set up its cancer-genome project in 2000, when sequencing was still a comparatively arduous business. They were interested in looking at mutations of 40 different genes, but practical limitations meant they could only start working on 20. The third gene they looked at was BRAF; sequencing the genes from 500 cancers the researchers found that there were BRAF mutants putting yet another cell-growth-signalling pathway into overdrive in half or more of the malignant melanomas in their sample. By 2011 the first BRAF inhibitor, Zelboraf (vemurafenib), was approved for the treatment of melanoma. In a trial, the six-month survival was 84%, compared with 64% who were treated with chemotherapy. The drug was quite toxic—despite their targeting, such drugs do have side-effects—but it was still approved for use.
Hundreds of thousands of cancers have now been sequenced, and the hunt for targets is seeing diminishing returns. Though there are hundreds of genes that go wrong in cancers, only a limited number promote cancer development and are common to a number of cancers. A significant amount of work is now focused not on finding new targets but on second-generation drugs aimed at targets that have already proved vulnerable; these newer drugs aim for higher efficacy, lower side effects or, ideally, both. There are also ways to combine the specificity of antibodies aimed at a well characterised target with other forms of treatment—to bind the antibody to something poisonous, say, or to something radioactive, and use it as an address label.
But there are still new targets being hit for the first time. In 2016, the drug Xalkori (crizotinib) was approved for ROS1-positive lung cancer. Louis Staudt, director of the centre for cancer genomics at the National Cancer Institute (NCI) in Bethesda, Maryland, says about 1-3% of cases of lung cancer are driven by a ROS1 mutation. Dr Staudt is working on a repository for genomic information called the NCI Genomic Data Commons, which hopes to identify more low-frequency drivers of cancer.
These targeted therapies are changing the way the doctors and regulators look at cancer. Typically cancers have been classified according to where they occur and how they behave. Now they can also be classified according to which genes are going wrong in them. This allows new sorts of investigation such as the NCI’s MATCH trial, which matches patients to treatments based on the genetic changes in their tumours. More than 6,000 patients treated at more than 1,000 institutions have had their tumours sequenced as part of this trial. The large numbers are needed to pick out the rare mutations that drive cancers.
By their genes shall you know them
A milestone in the transition to a genomic era for cancer therapy was reached earlier this year when America’s Food and Drug Administration (FDA) approved a treatment based on a specific genetic indicator rather than the type of tumour, as determined by location and its tissue structure. A similar “biomarker”-based approval is expected soon for a drug which targets a defect in a family of signalling proteins called tropomyosin receptor kinases, proteins which play an important role in tumour growth. A rare mutation (it affects only about 1% of patients) sees the genes that code for TRKs become fused to other genes. Loxo Oncology, a biotechnology firm in Stamford, Connecticut, has developed a drug aimed at this aberration; the idea is that it should be licensed for use in anyone with the relevant mutation.
Targeted therapies mark a significant advance over, and addition to, older chemotherapies. But they share their familiar weaknesses. In the 2000s Olivia Rossanese, a researcher at the ICR, worked on a BRAF inhibitor at the British pharma firm GSK. She says: “We made a drug to it, we said patients with this mutation are going to respond and that happened. It was a beautiful story …right up until resistance.” To cancers, targeted therapies, including antibodies, are another constraint to evolve around, and in the end that is what they tend to do.
For 13 months Ms Milley responded wonderfully to Tarceva. Then her doctor at the Dana-Farber Cancer Institute in Boston noticed spots on her adrenal glands that indicated resistance. That was the point at which she started cisplatin chemotherapy. It worked for eight months. When it started to falter, she went back on to the Tarceva, which worked for another four months. That looked like the end of the road for approved treatments. The only remaining option seemed to be to take a chance with a clinical trial, and this she agreed to do.
In the middle of October 2015 her doctor called unexpectedly. She was not eligible for the trial she had been trying to enroll in. But the FDA had just approved a brand new drug for lung cancer: Keytruda (pembrolizumab). It was one of a very promising new class of treatment known as immunotherapies.
Medicine has finally figured out how to get the immune system to fight cancer
THAT infections could sometimes cause a cancer to retreat, or even vanish, was known well before the advent of modern medicine. Imhotep, a pharaoh, recommended treating a tumour with a poultice followed by an incision—something that would help an infection develop. In early modern Europe doctors used septic dressings on tumours with ulcers and deliberately created purulent sores. By the end of the 19th century, William Coley, a bone surgeon in New York, was methodically infecting patients with Streptococcus bacteria.
Coley’s work fell out of favour, partly thanks to the rise of radiation therapy. Many continued to cling to the idea that the immune system might in some circumstances be provoked into recognising, attacking and subduing a cancer; they just didn’t know how to provide the provocation. In 1976 this latent belief in the potential of “immunotherapy” blossomed into hope with the discovery of interleukin 2 (IL-2). IL-2 is a growth factor that encourages the production of T-cells, white blood cells that scan the body for unwanted invaders and, on finding them, activate other parts of the immune system, including the B-cells which produce antibodies.
But IL-2 was a false dawn. On its own, it activated the immune system indiscriminately, and the immune system is a powerful beast; Elad Sharon, at the National Cancer Institute’s division of treatment and diagnosis, says the effects were “toxic and messy, and frequently sent patients to the ICU.” That might have been more tolerable in a drug that delivered. But to general surprise and discouragement IL-2 cured only a few patients of metastatic cancers. It was not clear why the treatment was not more effective.
Answers started to arrive in the 1990s. James Allison, at the Cancer Research Laboratory at the University of California, Berkeley, began work on a protein called CTLA-4 on the surface of some T-cells. By 1996 he had shown that this protein put a brake on the immune response to cancer. Blocking CTLA-4 with an antibody removed the brake; the immune system activated itself and got to work. Tumours in mice vanished when the animals were given CTLA-4-blocking antibodies. Though it was not immediately obvious, in retrospect this came to be seen as one of the reasons IL-2 never really worked: it is not possible to make a car run faster if its brakes are jammed on.

At the time oncologists were unimpressed by Dr Allison’s results. Cancer had been cured in mice many times over. And after many failed trials, immunotherapy was in exile—banished to the small corners of the big oncology meetings. But in 1999 Tasuku Honjo of the University of Kyoto, in Japan, showed that the gene for a protein called PD-1 also seemed to tamp down the immune system. When this gene was switched off in mice, some developed autoimmune diseases—a sign of an immune system in overdrive. In collaboration with Arlene Sharpe and Gordon Freeman at Harvard, Dr Honjo showed that some cancer cells had a second protein called PD-L1 on their surfaces which, by interacting with the PD-1 on T-cells, protected the cancer from them. Dr Honjo remembers approaching many companies with the finding, but “none wanted to invest.”
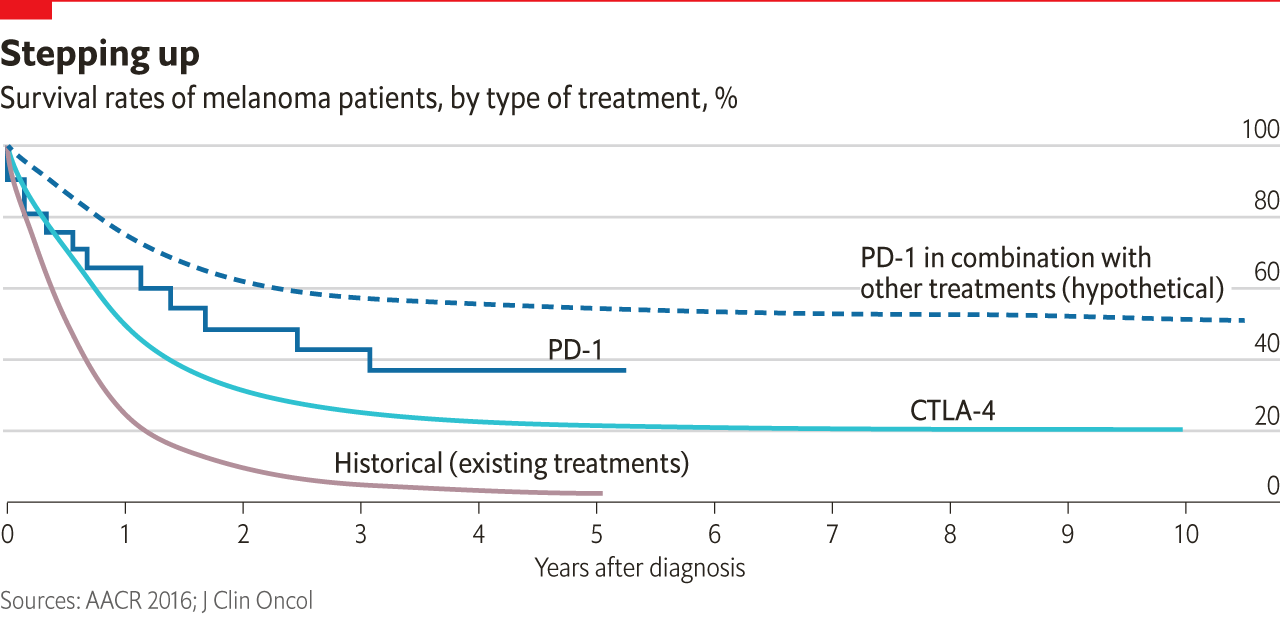
Despite a general wariness on the part of pharma companies, though, a trickle of development on therapies aimed at CTLA-4 and PD-1 did begin. Then, in 2010, Bristol-Myers Squibb released results from a trial of an anti-CTLA-4 antibody, Yervoy (ipilimumab), in malignant melanoma. Compared with the state of the art, they were fantastic. It was the first drug shown to change survival in this devastating disease, raising the median to ten months. Some survived much longer.
What was going on? Because the immune system is such a powerful beast, evolution has equipped it with a system of checks and balances lest it get out of hand. Both CTLA-4 and PD-1 are parts of that system. When one sort of immune cell presents an antigen which it has picked up to another, the second cell will ignore it if, at the same time, the first cell stimulates the CTLA-4 receptor. If the CTLA-4 receptor is blocked with an antibody like Yervoy, though, this “checkpoint” system does not work. Unchecked, the immune system is able to react to a wider range of antigens—including tumour antigens. Freed up by Yervoy the body’s T-cells started attacking the melanomas. And, it turned out, kept on attacking them. Perhaps the most remarkable feature of the new “checkpoint inhibitor” was that a small subset of patients survived for year after year.
Despite indications of success with melanomas, many scientists thought the checkpoint-inhibitor mechanism would not be broadly effective. Melanomas accumulate a very large array of mutations, and are thus more likely than most cancers to display antigens which trigger an immune response. This argument was bolstered by the observation that melanomas are more likely than other cancers to be subject to spontaneous remissions—presumably because something else kicks the immune system into gear. What was more, Yervoy had serious, sometimes life-threatening side-effects.
Pessimists have a pretty good record when it comes to cancer prognostication. But this time they were wrong. At Merck Roger Perlmutter, an immunologist who had previously left the company, was brought back to run the research labs. He became very interested in a PD-1-blocker then known only as MK-3475. Unlike CTLA-4, which works higher up the immune system’s chain of command, PD-1 has a front-line role; if a cancer cell carries PD-1’s counterpart, PD-L1, on its surface, T-cells will ignore the cell despite any suspicious antigens it may be carrying (see diagram). MK-3475 seemed to block the interaction nicely. It might thus render the immune system blind to the cancer’s subterfuge. “Whatever [else] you are doing, stop,” Perlmutter told his clinical-development group. Merck expanded a phase 1 trial programme looking at the drug’s effect on advanced melanoma to more than 1,200 patients, making it the largest phase 1 trial in the history of oncology.


The expansion was in part a response to a new discovery: early evidence suggested that checkpoint inhibitors could also get results with lung cancers, which are a much bigger killer than, and thus represent a much larger market than, malignant melanomas. Luis Diaz, head of solid-tumour oncology at the Memorial Sloan Kettering Cancer Centre in New York, recalls: “It was completely unexpected. Prior to that I was not a believer in immunotherapy.”
Whispers of a cure
Merck’s PD-1 drug would eventually be given the commercial name Keytruda (pembrolizumab). In 2014 it became the second checkpoint inhibitor to be approved in America—the world’s largest and most lucrative pharmaceutical market. Opdivo (nivolumab), a PD-1 drug which Ono Pharmaceuticals had developed on the basis of Dr Honjo’s work, soon joined it, having been licensed in Japan a little earlier. In some cases the drug produced effects little short of miraculous. In 2016 it was announced that it had cleared former president Jimmy Carter of metastatic melanoma that had spread to his liver and brain.
In lung cancer, and in many other cancers, the patients who responded tended to have a higher mutational burden, like that seen in melanoma. More antigens means more targets for the immune system to tackle when the drug lets it off the leash. This observation provided a way to spot some of the patients most likely to benefit. In 2017 Keytruda was approved for use in any cancer that has mismatch-repair-gene defects, a flaw which means that a cancer accumulates even more mutations—and thus more possible antigens—than most.
Another indicator that the drug may have something to offer is the tumours’ expression of PD-L1. Tumours expressing a lot of PD-L1 are investing in keeping the immune system duped; when the PD-1 system is interrupted they should prove particularly vulnerable. At the start of October 2015, Keytruda was approved for use in advanced non-small-cell lung cancer in cases where other treatments had failed and when there was PD-L1 on more than 50% of the tumour cells. Ms Milley’s score was 80%, and she started treatment almost immediately.
Jedd Wolchok, a medical oncologist at Memorial Sloan Kettering, says immunotherapies do not have the same kinds of impact as other types of cancer therapy. In some cases they do not work at all. In other cases they can either eliminate the cancer entirely, or cause it to stabilise, or regress. Responses to therapy are often longer lasting than those seen in targeted drugs. And they tend to persist after patients stop taking the drug (at present CTLA-4 drugs are usually administered for only a matter of months).
The nature of the long-lasting responses is intriguing. Dr Wolchok has patients who started treatment for malignant melanoma eight years ago. He finds it particularly interesting that in some cases scans of the cancers taken before treatment (when the prognosis for the patients would have been six or seven months) and scans taken today look more or less equally dreadful. Biopsies of the tumours reveal a lot of immune cells and a lot of dead tumour cells. Dr Wolchok says it looks like a “chronic struggle between a patient’s immune system and cancer”. This apparent equilibrium is quite different from what is seen in chemotherapy, where the cancer will be either susceptible or resistant. The difference seems to be due at least in part to the fact that the immune response, like the cancer, can evolve.
Though immunotherapy is still new, it has already radically shifted the treatment and research landscape. A wide range of combinations is being tested in the hope of improving patients’ responses. A trial combining Opdivo and Yervoy in malignant melanoma has shown tumours to shrink in 60% to 70% of patients (although it causes serious side-effects). Dr Wolchok says it is not yet possible to calculate the median survival time in the trial population—because more than half of the patients are still alive.
Compared with the more limited range of patients that can be treated with most targeted therapies, immunotherapies seem to work in many cancers. And as Dr Sharon at the NCI points out, it also produces cures. But this excitement needs to be tempered with the grittier reality that, across all cancers tried so far, only about 20% respond to the new approach. The response varies greatly between types of cancer. In patients who have failed the usual treatments for Hodgkin’s lymphoma it is 90%. In pancreatic and most colorectal cancers it is basically zero.
Improving this response is perhaps today’s biggest oncological challenge—the source of more excitement, and investment, than any other recent development in the field. Part of the answer will come from a better understanding of the steps needed to generate an anti-tumour response from the immune system, and of the therapeutic targets available. For example, Hervé Hoppenot, the boss of biotech firm Incyte, a biotech firm based in Wilmington, Delaware, says that some tumours protect themselves from the immune system using another checkpoint, IDO1 (an enzyme that was first discovered in a search for ways to protect a fetus from immune rejection). Incyte is testing epacadostat, an existing drug known to inhibit IDO1, as a cancer treatment both alone and in combination with PD-1 blockers.
There are well over 1,000 clinical trials of checkpoint inhibitors going on; what was at first a trickle, then a current, is now a torrent. Some worry that things have gone too far too fast. Jeff Bluestone, who runs the new Parker Institute for Cancer Immunotherapy in San Francisco, says “many of [these trials] are based on minimal data and very limited clinical evidence about what combination will work”. Some fear there are too few patients to allow these trials to be run, others that there is too little thought and planning and a lot of duplication of effort. Dr Freeman at Harvard says he has been told there are over 80 Chinese groups developing different PD-1 antibodies.
This enthusiasm may lead to wasted efforts, and even delay progress. But there is no doubt that immunotherapy will from here on be a key part of treatment for a growing number of cancers. Perhaps the most telling measure of its success is that some oncologists have started to complain, quietly, of a shortage of specialist doctors. Patients keep coming back instead of dying.

There is a lot more for immunotherapy to do
IMMUNOTHERAPY offers huge promise, both as an addition to established therapies and as a foundation for future ones. Hundreds of trials are pairing CTLA-4, PD-1 or PD-L1 inhibitors with chemotherapy, radiation and targeted therapies. One hope is that the older treatments will increase the range of antigens that the cancer offers for the immune system to latch on to, both by driving further mutations and by killing cancer cells. Dead cells release more antigens,
Then there is the development of further immunotherapies, which is being pursued both by building on the successes of the first checkpoint inhibitors and by using entirely new technologies, such as genome editing. Dr Wolchok at Memorial Sloan Kettering is working on the next generation of immune-modulators. These include new inhibitory compounds for IDO and TIM-3, another checkpoint. Some researchers are trying to remove further brakes on the system by killing or silencing some of its regulatory cells. Others are looking at molecules which activate the immune system in a similar way to IL-2. Nektar Therapeutics, a biotech firm based in San Francisco, is developing an engineered therapy which does this in a way that should, in principle, encourage tumour-killing T-cells. It is being tested as a combination treatment with an anti-PD-1 drug in five tumour types, including bladder cancer and a hard-to-treat form of breast cancer.
Other approaches seek to make sure that the immune system responds to as many cancer antigens as possible. Viruses genetically engineered to attack cancer cells might be used to this end. Even if such viruses did not kill enough cells to do the cancer much damage, the way in which they kill the cells would release otherwise hard-to-detect antigens that might help the immune system target the tumour better.
Alternatively, the antigens could be provided from outside. Now that immunotherapies have wind in their sails, various old ideas are coming back into vogue. One of them is vaccination. The vaccines with which people are familiar are those against infectious disease. They work by priming the immune system to respond to an antigen associated with a specific pathogen, so that when the system encounters the infection for real it already knows how to fight it. Because some infections can lead to cancer, some of these vaccinations can prevent it. Sometimes, as in vaccination against hepatitis B, which can cause liver cancer, this is an added bonus. Sometimes, as in vaccination against human papilloma virus, which can cause cervical cancer, it is the main point.
But there may be another way to use vaccines against cancer. Equipped with the right antigen, a vaccine might encourage an immune response to a tumour which is already present, but which the immune system has failed to get to grips with. It is an approach that has been frequently tried in the past, and has repeatedly failed. But the availability of checkpoint inhibitors and the ability to pick out the most promising antigens may allow this form of targeting to come into its own.
Neon Therapeutics, Gritstone Oncology, Genocea Biosciences and other biotech firms are all pursuing the creation of personalised vaccines based on the mutations in an individual tumour. The trick is to find which of the novel antigens its genome says the tumour might be churning out are the most likely to provoke a strong response when served up to the immune system in the form of a vaccine. Jill O’Donnell-Tormey at the Cancer Research Institute (CRI), a non-profit in New York that concentrates on immunotherapies, says that everyone has their favourite algorithm to predict which antigens will get the best response. Together with the Parker Institute in San Francisco, CRI is creating a “bake off” where these algorithms will be tested against each other.
If vaccines work in late-stage cancer—which is where most therapies are tried first—there might be scope for bringing them in sooner, at least in some cancers. In decades to come it is possible to imagine an approach where a tailored vaccine might be the first—and, ideally, the only—response to a blood test showing the presence of a cancer.
Reprogramming the genome
Like immunotherapies, vaccines offer a way to hack the immune system by changing the way that its cells fight the cancer and increasing the number of them doing so. A less circuitous way of doing this is now on offer: reprogram the immune system directly. Take some of its cells out of the body, manipulate them so that they do what you want, encourage them to divide and multiply, then put them back and let them get on with the job.
The technology along these lines that has got furthest is called CAR-T, where CAR stands for “Chimeric antigen receptor”. These CARs are produced by splicing together the gene for an antibody that recognises a tumour antigen and the gene for a receptor that sits on the surface of the T-cells; put this new gene into a T-cell and it will be precisely targeted at the tumour. The small clinical trials undertaken to date suggest that this could be extremely effective. A trial of 31 patients with acute lymphoblastic leukaemia brought a complete, and unprecedented, remission in 93% of cases. A CAR-T therapy called Kymriah (tisagenlecleucel), made by the Swiss firm Novartis to treat B-cell acute lymphoblastic leukaemia, was approved for use in America on August 30th.
There are two main limitations to CAR-T. One is that so far the T-cells have been programmed to target a molecule, CD19, which is only common to the surface of a few blood cancers. The other is that CAR-T has been known to trigger immune reactions which can prove fatal. Neither problem is obviously insoluble. Editing genes has been made much easier by a new technology known as CRISPR-Cas9, which has already been used to improve the way that CAR-T cells are engineered in mice. It may well eventually allow the receptors used in such therapies to be personalised to the specifics of the patient’s cancer. And more precision, as well as experience, should lead to immune responses less likely to run away with themselves.
What such advances will not do, though, is make such treatments cheaper. Novartis’s new therapy costs $475,000. Genome-editing treatments seem likely to be the most expensive cancer treatments the world has yet seen. And that is saying quite a lot, since many of the newer cancer treatments are eye-wateringly pricey (see chart ).
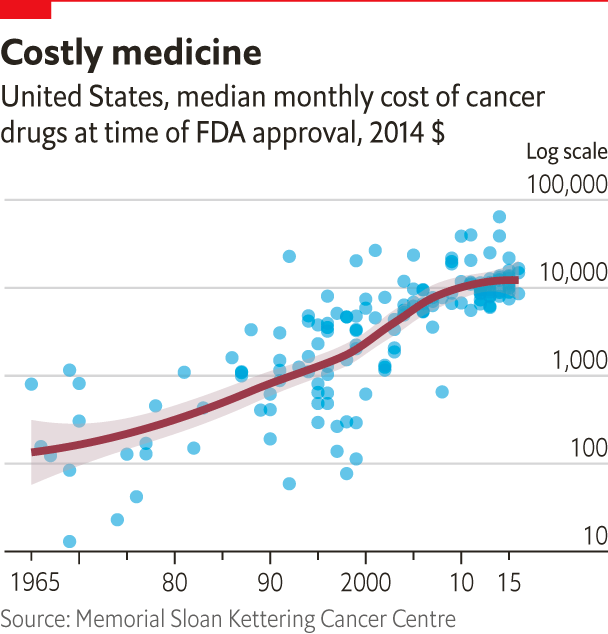
There are various reasons for this. More sophisticated R&D costs a lot. And antibodies are much more expensive to make than the smaller molecules used in older therapies. Generic versions of them are still few and far between. A company than can make antibodies which pass regulatory muster is much better advised to make ones it can sell for a premium.
But the overwhelming factor is that in America, the world’s largest market for drugs, prices are set by what the market will bear. When life-saving drugs are available from only one or two providers high prices are a given. This is why pharma companies have piled into oncology over the past decade. They see a market which, by 2025, is forecast to be worth $45bn-100bn a year.
Not all progress is expensive. Effective early diagnostics could save both money and suffering. The knowledge gained from blood biopsies should allow doctors to tailor treatments better, and avoid drugs that will not work on a given patient. And in a different economic setting bespoke vaccines, gene-editing treatments and the like could in times to come short-circuit rising prices. Molecules made inside the body by reprogrammed cells should be cheaper than those made in expensive cultures. Cutting and splicing the genome could be a great deal cheaper than using scalpels and lasers on the body.
But in the world as it is new cancer therapies will continue to be among the most expensive interventions medicine has to offer, creating a challenge for health systems around the world. And some will disappoint. The immune system’s complexity means that it will not always react as doctors hope. Some treatments will prove less effective than at first they seemed. This is a particular problem for cancer drugs, which tend to be approved after comparatively small trials. A recent study of 36 drugs approved between 2008 and 2012 found that 18 did not help patients to live longer. The price of these drugs ranged from $20,000 to almost $170,000 per patient.
The incidence of cancer will continue to be dominated by demographics. In developed countries, new therapies may not reduce the chances of getting cancer for some time, simply because older people get more cancers. But the chances of surviving your first cancer, or your next cancer, will improve—and for those with more amenable cancers, and access to the best treatment, they may do so quite quickly. Ever more people will still be told, “I’m afraid you have cancer.” But the words will become less fateful, the diagnosis ever less feared.
The cost of progress
When Ms Milley was diagnosed with advanced lung cancer, she went on to Google and read the words “death sentence”. It is, alas, fairly typical for patients with terminal cancer to have little idea about their prognosis unless they seek it out. Many might be better served by more openness.
But prognosis is not destiny. Ms Milley started taking Keytruda in December 2015. After two months of treatment her lesions had almost entirely vanished. So far, they have mounted no comeback, and she continues to feel well. She finds the response “amazing”.
On any given drug, in any given trial, most people will not be as fortunate. But one of the strange consolations of the current progress being made against cancer is that modern biomedicine makes it possible to learn more from failure than ever before. Huge amounts of the knowledge now saving lives was gained from dead and dying patients, loved ones and friends who lost their fight for life but left a legacy of data. In any given case, that is scant recompense. Put those contributions together, though, and they make a remarkable memorial.